


Hot Topics #24 (May 27, 2024)
Flow matching for protein ensembles, pLM guided protein fitness prediction, LLM interpretability, and more.

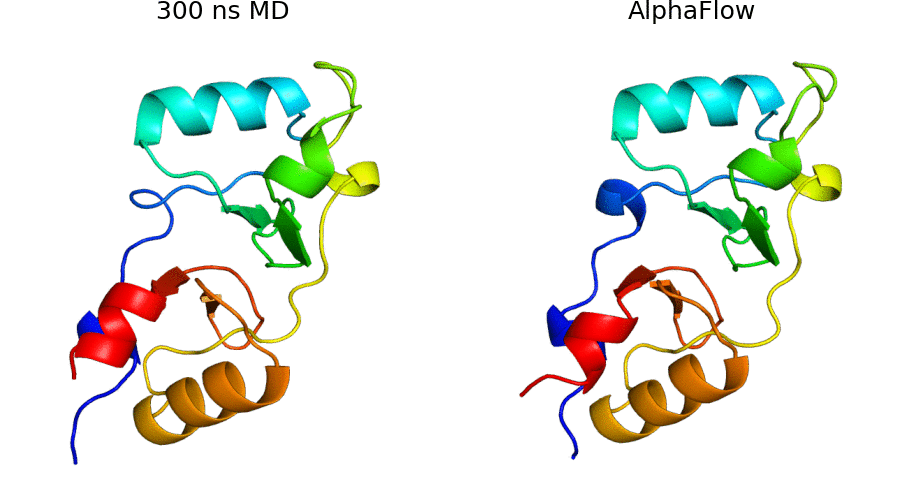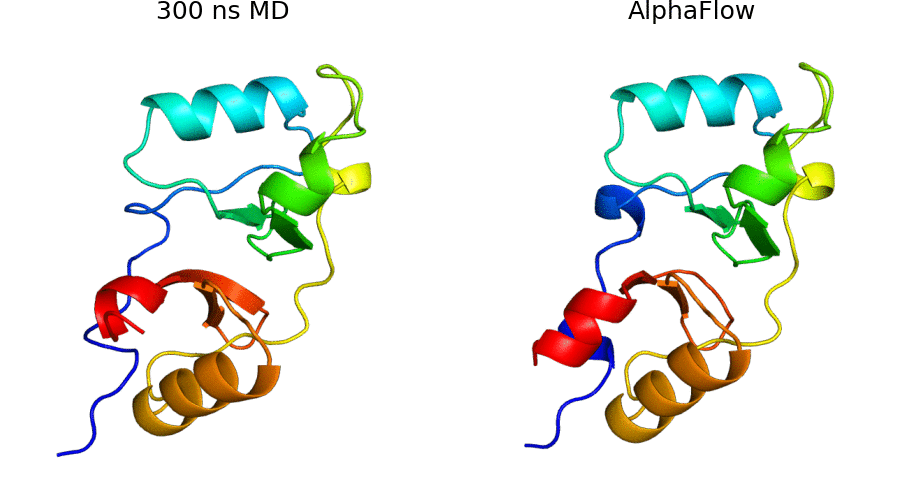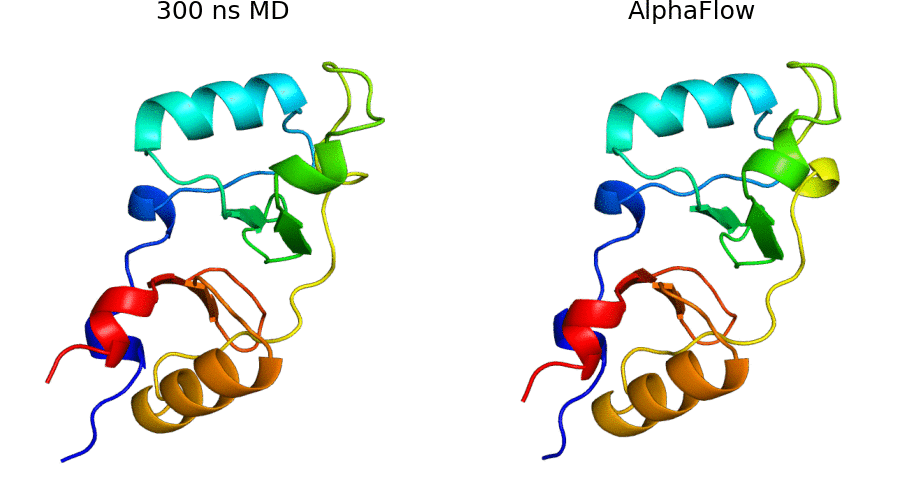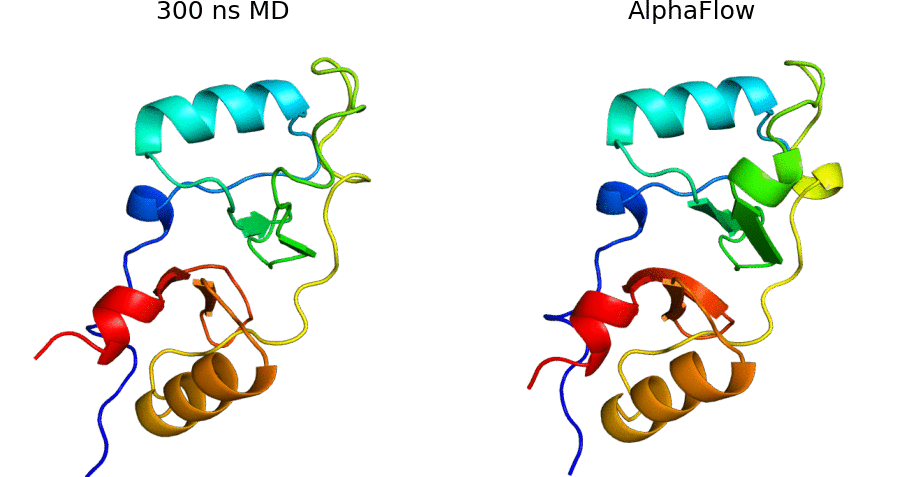

AlphaFold Meets Flow Matching for Generating Protein Ensembles: Jing et al.: Feb 7, 2024
Abstract: The biological functions of proteins often depend on dynamic structural ensembles. In this work, we develop a flow-based generative modeling approach for learning and sampling the conformational landscapes of proteins. We repurpose highly accurate single-state predictors such as AlphaFold and ESMFold and fine-tune them under a custom flow matching framework to obtain sequence-conditoned generative models of protein structure called AlphaFlow and ESMFlow. When trained and evaluated on the PDB, our method provides a superior combination of precision and diversity compared to AlphaFold with MSA subsampling. When further trained on ensembles from all-atom MD, our method accurately captures conformational flexibility, positional distributions, and higher-order ensemble observables for unseen proteins. Moreover, our method can diversify a static PDB structure with faster wall-clock convergence to certain equilibrium properties than replicate MD trajectories, demonstrating its potential as a proxy for expensive physics-based simulations. Code is available at this https URL.
VespaG: Expert-guided protein Language Models enable accurate and blazingly fast fitness prediction: Marquet et al.: April 28, 2024
Abstract: Exhaustive experimental annotation of the effect of all known protein variants remains daunting and expensive, stressing the need for scalable effect predictions. We introduce VespaG, a blazingly fast single amino acid variant effect predictor, leveraging embeddings of protein Language Models as input to a minimal deep learning model. To overcome the sparsity of experimental training data, we created a dataset of 39 million single amino acid variants from the human proteome applying the multiple sequence alignment-based effect predictor GEMME as a pseudo standard-of-truth. Assessed against the ProteinGym Substitution Benchmark (217 multiplex assays of variant effect with 2.5 million variants), VespaG achieved a mean Spearman correlation of 0.48±0.01, matching state-of-the-art methods such as GEMME, TranceptEVE, PoET, AlphaMissense, and VESPA. VespaG reached its top-level performance several orders of magnitude faster, predicting all mutational landscapes of the human proteome in 30 minutes on a consumer laptop (12-core CPU, 16 GB RAM).
Availability VespaG is available freely at https://github.com/JSchlensok/VespaG
Large scale analysis of predicted protein structures links model features to in vivo behaviour: Stam et al.: April 14, 2024
Abstract: Rapid advancements in protein structure prediction methods have ushered in a new era of abundant and accurate structural data, providing opportunities to analyse proteins at a scale that has not been possible before. Here we show that features derived solely from predicted structures can be used to understand in vivo protein behaviour using data-driven methods. We found that these features were predictive of in vivo protein production for a set of designed antibodies, enabling identification of high-quality designs. Following on from this result, we calculated these features for a diverse set of ≈500,000 predicted structures, and our analysis showed systematic variation between proteins from different organisms to such an extent that the tree of life could be recapitulated from these data. Given the high degree of functional constraint around the chemistry of proteins, this result is surprising, and could have important implications for the design and engineering of novel proteins.
Scaling Monosemanticity: Extracting Interpretable Features from Claude 3 Sonnet: Templeton et al.: May 21, 2024
Abstract: Eight months ago, we demonstrated that sparse autoencoders could recover monosemantic features from a small one-layer transformer. At the time, a major concern was that this method might not scale feasibly to state-of-the-art transformers and, as a result, be unable to practically contribute to AI safety. Since then, scaling sparse autoencoders has been a major priority of the Anthropic interpretability team, and we're pleased to report extracting high-quality features from Claude 3 Sonnet, 1 Anthropic's medium-sized production model.
We find a diversity of highly abstract features. They both respond to and behaviorally cause abstract behaviors. Examples of features we find include features for famous people, features for countries and cities, and features tracking type signatures in code. Many features are multilingual (responding to the same concept across languages) and multimodal (responding to the same concept in both text and images), as well as encompassing both abstract and concrete instantiations of the same idea (such as code with security vulnerabilities, and abstract discussion of security vulnerabilities).
State-specific protein-ligand complex structure prediction with a multi-scale deep generative model: Qiao et al.: Sep. 30, 2022
Abstract: The binding complexes formed by proteins and small molecule ligands are ubiquitous and critical to life. Despite recent advancements in protein structure prediction, existing algorithms are so far unable to systematically predict the binding ligand structures along with their regulatory effects on protein folding. To address this discrepancy, we present NeuralPLexer, a computational approach that can directly predict protein-ligand complex structures solely using protein sequence and ligand molecular graph inputs. NeuralPLexer adopts a deep generative model to sample the 3D structures of the binding complex and their conformational changes at an atomistic resolution. The model is based on a diffusion process that incorporates essential biophysical constraints and a multi-scale geometric deep learning system to iteratively sample residue-level contact maps and all heavy-atom coordinates in a hierarchical manner. NeuralPLexer achieves state-of-the-art performance compared to all existing methods on benchmarks for both protein-ligand blind docking and flexible binding site structure recovery. Moreover, owing to its specificity in sampling both ligand-free-state and ligand-bound-state ensembles, NeuralPLexer consistently outperforms AlphaFold2 in terms of global protein structure accuracy on both representative structure pairs with large conformational changes (average TM-score=0.93) and recently determined ligand-binding proteins (average TM-score=0.89). Case studies reveal that the predicted conformational variations are consistent with structure determination experiments for important targets, including human KRASG12C, ketol-acid reductoisomerase, and purine GPCRs. Our study suggests that a data-driven approach can capture the structural cooperativity between proteins and small molecules, showing promise in accelerating the design of enzymes, drug molecules, and beyond.
Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies: Ruffolo et al.: April 25, 2023
Abstract: Antibodies have the capacity to bind a diverse set of antigens, and they have become critical therapeutics and diagnostic molecules. The binding of antibodies is facilitated by a set of six hypervariable loops that are diversified through genetic recombination and mutation. Even with recent advances, accurate structural prediction of these loops remains a challenge. Here, we present IgFold, a fast deep learning method for antibody structure prediction. IgFold consists of a pre-trained language model trained on 558 million natural antibody sequences followed by graph networks that directly predict backbone atom coordinates. IgFold predicts structures of similar or better quality than alternative methods (including AlphaFold) in significantly less time (under 25 s). Accurate structure prediction on this timescale makes possible avenues of investigation that were previously infeasible. As a demonstration of IgFold’s capabilities, we predicted structures for 1.4 million paired antibody sequences, providing structural insights to 500-fold more antibodies than have experimentally determined structures.

Biotite: Biotite is your Swiss army knife for bioinformatics. Whether you want to identify homologous sequence regions in a protein family or you would like to find disulfide bonds in a protein structure: Biotite has the right tool for you. This package bundles popular tasks in computational molecular biology into a uniform Python library. It can handle a major part of the typical workflow for sequence and biomolecular structure data
AlphaFlow code: AlphaFlow is a modified version of AlphaFold, fine-tuned with a flow matching objective, designed for generative modeling of protein conformational ensembles.
VespaG code: VespaG is a blazingly fast single amino acid variant effect predictor, leveraging embeddings of the protein language model ESM-2 (Lin et al. 2022) as input to a minimal deep learning model.